工作流操作
本文旨在介绍GeneDock平台中工作流(Workflow)的具体操作方法,主要包括如何创建和运行工作流。
工作流的分类:
- “公共工作流”:系统配置好的工作流,供大家直接使用,但用户无权限编辑。公共工作流可以通过点击“公共库” –> “公共工作流”来添加。(公共工作流列表详见下方附录)
- “自有工作流”:用户自己创建的工作流,在GeneDock平台上运行任务,用户有权进行编辑。
如何创建工作流?
创建工作流就是将你的工具串连起来的过程。
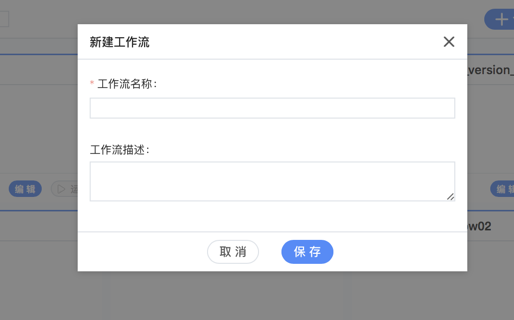
填写名称和描述
- 点击GeneDock平台上“工作流” –> “创建工作流”;
- 在跳出弹窗中,填写“工作流名称”和“描述”信息;
- 点击“保存”按钮。
选择工具
- 在左侧“工具列表”中,点击添加任意一个工具(包括系统、公共和自有工具);
- 添加工具会有一个配置弹窗,用户可以选择工具版本、工具别名(alias)、参数默认值和参数值是否可更改;
注意,加载文件需要添加系统工具“loaddata”,保存文件需要添加系统工具“storedata”。

串连工具节点
添加工具后,生成一个节点,可以根据工具间的输入和输出关系,将这些节点串起来,形成一个有向无环图(DAG)。
操作步骤如下:
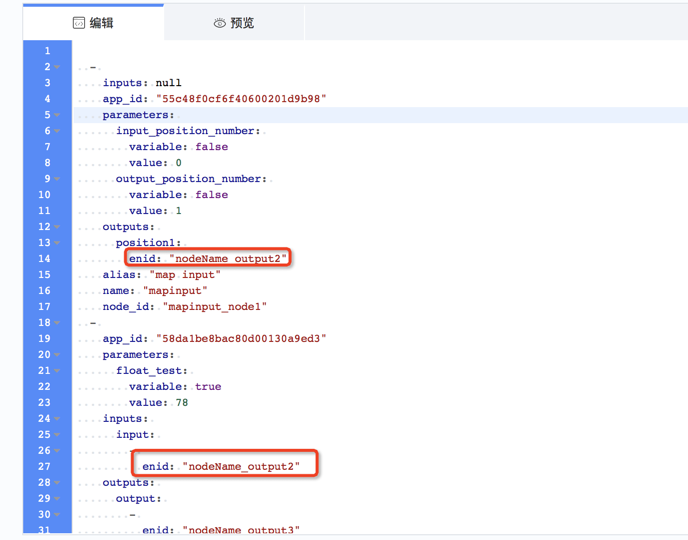
- 添加工具后,”编辑”窗口新增一段代码块。代码块包含工具的输入项、输出项和参数项,以及
alias别名项和node_id节点名称; node_id是节点的唯一标识,如有保存工作流的时候会报错;alias来命名节点的别名,方便”预览”的时候查看;inputs是节点的输入项,用来对接上一步节点的outputs,系统工具loaddata和mapinput的inputs是null;outputs是节点的输出项,用来对接下一步节点的inputs,系统工具storedata和mapoutput的outputs是null;enid是表示输入、输出项的名称。用户可用enid来连接两个节点,即将节点1的输出项的enid和节点2的输入项的enid设为同样的名称。- 用户可切换到“预览”标签,查看节点的预览效果
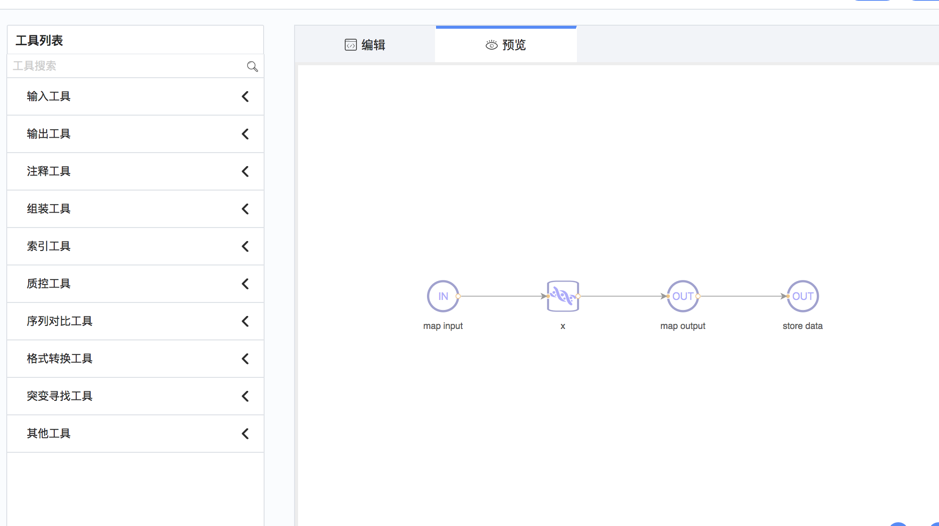
保存工作流
- 点击右上角“保存”按钮;
- 用户可以对“描述”再次更改,如不改变本工作流的版本,选择覆盖本次更新,否则系统会新增工作流版本。
- 点击右下角“确定”按钮。

如何编辑工作流?
- 在“我的工作流”页面,找到对应工作流,点击“编辑”按钮;
- 更改工作流相应配置;
- 点击“保存”按钮,保存配置更改,如不改变本工作流的版本,选择覆盖本次更新,否则系统会新增工作流版本。
- 点击“另存为”按钮,可以将当前的工作流,保存成其他名字的工作流;

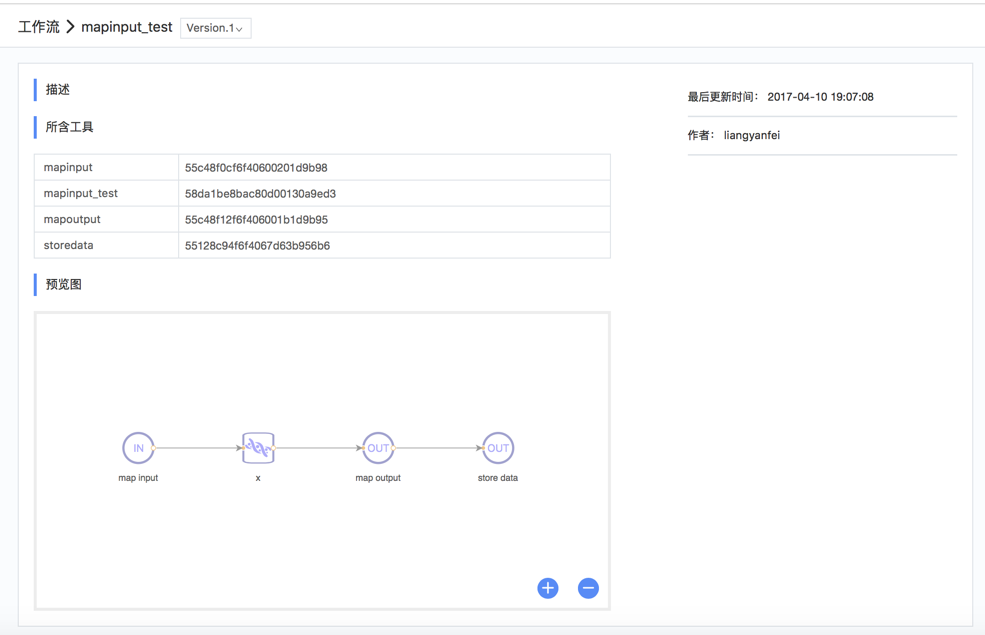
查看工作流详情
在“我的工作流”页面,找到对应工作流,点击该工作流卡片即可查看“基本信息”、“描述”、“所含工具”和“预览图”信息。
如何运行工作流?
- 在“我的工作流”页面,找到对应工作流,点击“运行”按钮;
- 设置输入文件,设置输出文件路径,设置参数项配置等;
- 直接点击”运行任务”或者先”预览”再点击”运行”;
- 另外还可以将此次配置保存成默认配置,供下次该工作流运行时使用。
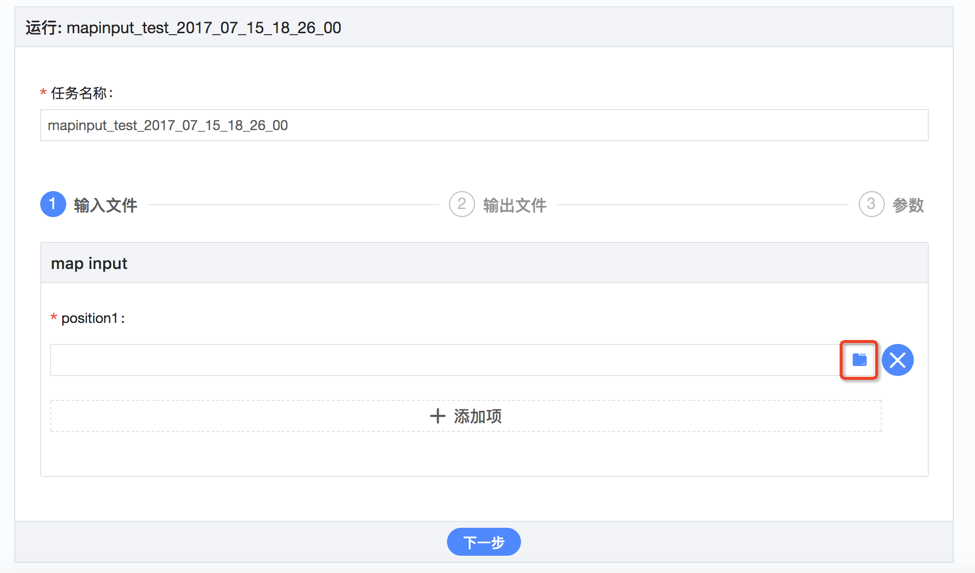
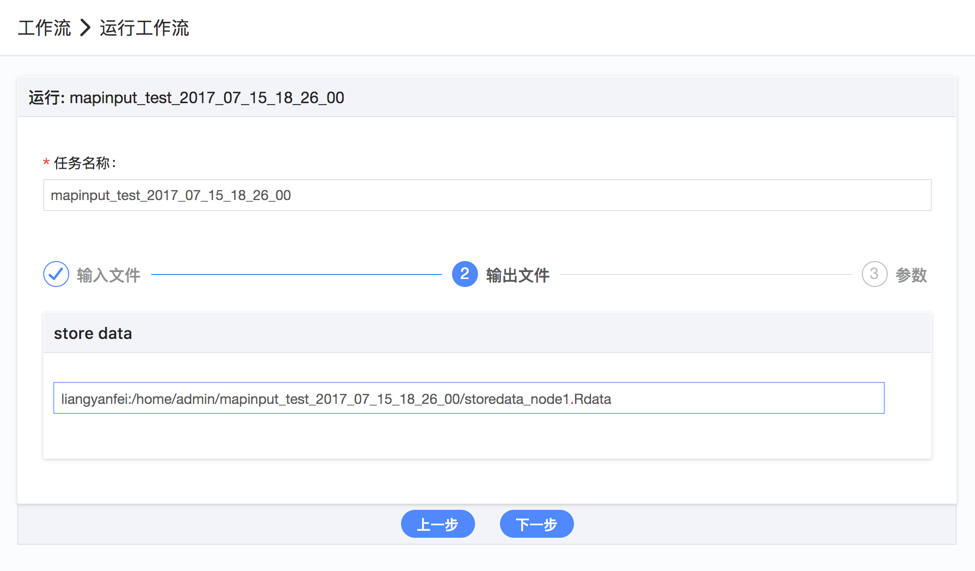
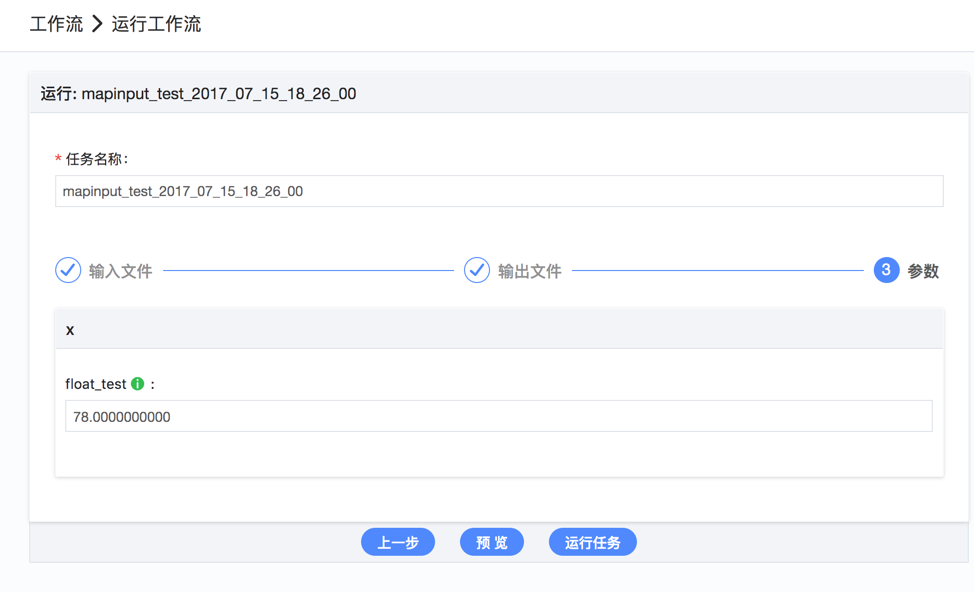
运行工作流会提交一个任务,你可以通过“任务/报告”查看任务的运行状态、参数、日志和报告。详见任务操作。
一些小贴士:
- 运行工作流时的输入文件、输出文件、参数的各个选项,均为工作流中工具时设置,并在创建工作流时设置的。
- 运行工作流时参数的默认值可以是工具中设置的,也可以是工作流创建时设置的。
- 运行工作流时参数的默认值是否可以被更改,主要是在创建工作流,添加工具时设置的“variable”参数。
- 对工具进行了更改,相应的工作流不会自动同步。因此,若工具的更改涉及输入、输出和参数项,您需要重新配置工作流对应部分;若工具的更改不涉及输入、输出和参数项,您需要点击“编辑工作流”,重新点击“保存”,以更新相应配置。
- 当下一步工具的一个输入项需要接上一步多个工具的输出项,且上一步的工具不包含loaddata时,您可以通过手动编辑,并列enid实现。(示例如下)
inputs:
read1:
- enid: "enid1"
- enid: "enid2"
附录
公共工作流列表(共7个)
| 序号 | 工具名称 | 备注 |
|---|---|---|
| 1 | Fastqc | |
| 2 | VEP_Ensembl_annotation | |
| 3 | VEP_RefSeq_annotation | |
| 4 | WES_Germline_BWA-GATK | 需自行上传GATK jar包 |
| 5 | WES_Germline_BWA-GATK4 | |
| 6 | WES_Somatic_BWA-GATK-Mutect1-Strelka | 需自行上传GATK jar包 |
| 7 | WGS_Germline_BWA-GATK | 需自行上传GATK jar包 |
| 8 | WGS_Germline_BWA-GATK4 | |
| 9 | Metagenome_Prinseq-BMtagger-HUMAnN2 |
工作流配置示例
此为工作流“编辑”页面配置示例,将公共工具“Bwa_mem”串成了一个自有工作流。
###一个node_id对应一个工具
###app_id为工具对应在系统中保存的id
###alias名字方便我们记忆
###enid需要手工修改,同时将输入与输出串起来
-
inputs: null
name: "loaddata"
parameters: null
outputs:
data:
enid: "loaddata_read1"
app_id: "55128c58f6f4067d63b956b5"
alias: "输入read1序列"
node_id: "loaddata_node1"
-
inputs: null
name: "loaddata"
parameters: null
outputs:
data:
enid: "loaddata_read2"
app_id: "55128c58f6f4067d63b956b5"
alias: "输入read2序列"
node_id: "loaddata_node2"
-
inputs: null
name: "loaddata"
parameters: null
outputs:
data:
enid: "loaddata_bwaindex"
app_id: "55128c58f6f4067d63b956b5"
alias: "输入含bwa索引的参考tgz文件"
node_id: "loaddata_node3"
-
inputs:
read1:
-
enid: "loaddata_read1"
read2:
-
enid: "loaddata_read2"
refgenome:
-
enid: "loaddata_bwaindex"
name: "Bwa_mem"
parameters:
read_group:
variable: false
value: " "
outputs:
outbam:
-
enid: "bwa_mem_outbam"
app_id: "575535945346800019fa9b8b"
alias: "bwa mem公共工具"
node_id: "Bwa_mem_node4"
-
inputs:
data:
enid: "bwa_mem_outbam"
name: "storedata"
parameters:
description:
variable: false
value: " "
name:
variable: false
value: " "
outputs: null
app_id: "55128c94f6f4067d63b956b6"
alias: "输出比对后的bam文件"
node_id: "storedata_node5"