引子
为了完成主任交代的任务–同时提交成百上千次分析,小F找到了肿瘤科的小W求助。
小F:“小W,上回咱俩交流完,我通过Python SDK已经成功在GeneDock平台提交了任务。我们主任为此还夸奖了我,好激动。可是我们那边样本量很大,经常是成百上千个,如何通过Python SDK批量提交这么多分析任务呢?一个一个地手动修改配置文件好辛苦,还容易出错。”
小W:“这个简单!这种情况的批量提交任务就相当于,按照样本信息把配置文件中的输入项和输出项批量替换,每次替换完配置文件提交一个任务。下面我给你详细说一下具体步骤:”
1. 获取运行任务的配置文件
小F:“上次我学会了如何修改运行任务相关的配置文件,那么这个配置文件是如何拿到的呢?”
小W:“这个配置文件其实就是可执行的工作流的参数模板。只需修改 gd_get_param.py 脚本中的2处,然后在Shell中运行 python gd_get_param.py > Fastqc_param_test.yml 命令即可。Fastqc_param_test.yml 就是获取的配置文件。”
(Python参考脚本1:gd_get_param.py)
#!/usr/bin/python
# -*- coding: utf-8 -*-
import gdpy
# 填写秘钥信息及ENDPOINT(此处需修改-1)
ACCESS_KEY_ID = ''
ACCESS_KEY_SECRET = ''
ENDPOINT = 'cn-beijing-api.genedock.com' # 以北京为例
# 工作流所属的资源账号,如果是公共工作流,此处应为'public'(此处需修改-2)
WORKFLOW_NAME = 'Fastqc'
WORKFLOW_VERSION = 1
RES_ACCOUNT_NAME = 'public'
# 初始化Auth信息
auth = gdpy.GeneDockAuth(ACCESS_KEY_ID, ACCESS_KEY_SECRET)
# 创建一个workflow对象
workflow = gdpy.Workflows(auth, ENDPOINT, RES_ACCOUNT_NAME)
# 获取某个可执行的workflow参数模板
resp = workflow.get_exc_workflow(WORKFLOW_NAME, WORKFLOW_VERSION)
# 将获取参数模板信息转换成yaml格式并输出
workflow_param = gdpy.yml_utils.yaml_dumper(resp.parameter)
print workflow_param
说明:
目前,也可以通过安装 gwc 客户端 (GeneDock Wofkflow Client) ,运行 “gwc workflow getparam” 获取运行任务的配置文件,其使用方法参考 GeneDock Workflow Client手册。
2. 修改运行任务的配置文件
小W:”上次运行任务时,你把配置文件的输出项和输出项直接修改成了实际文件路径信息。现在,把输入项和输出项用变量替换一下,根据样本名称和路径,用脚本批量替换这些需要修改的地方即可。下面我以公共工作流Fastqc(版本号:1)为例详细讲解一下:“
(1) 修改输入项:
将输入项的enid处替换成null,name处替换成变量。
例如:本例中只有一个输入项 “loaddata_reads”,将enid处的 < Please input the enid of the data in here > 替换成null,将name处的 < Please input the name of the data in here > 替换成 “{$read}” 。
注意:
1)如果引入了特殊符号$或{}等,一定要添加单引号或双引号,即 '{$read}' 或 "{$read}" 形式。
2)如果流程有多个输入项:
[1] 可以根据实际情况替换成不同的名称(注意:保证唯一性,方便利用Python脚本进行识别和替换);
[2] 有的输入项不需要根据样本名称进行变动,可在模板中填写输入项文件的实际路径,后续将不对这些输入项进行替换。
(2) 修改输出项:
将输出项的name处替换成变量。
例如:本例中只有一个输出项 “store_data_node_fastqc”,将name处的 < Please input the name of the output data in here > 替换成 “/home/admin/Fastqc_Result/{$sample}_fasqtc_out.tgz” 。其中,Fastqc_Result 为自己定义的输出文件夹名称,{$sample}_fasqtc_out.tgz 为根据样本名称自己定义的输出结果文件名。
注意:
1)根据Outputs的formats正确填写输出项的文件格式。
2)如果引入了特殊符号$或{}等,一定要添加单引号或双引号,即 '/home/admin/Fastqc_Result/{$sample}_fasqtc_out.tgz'
或 "/home/admin/Fastqc_Result/{$sample}_fasqtc_out.tgz" 形式。
3)如果流程有多个输出项:
可以根据实际情况替换成不同的名称(注意:保证唯一性,方便利用Python脚本进行识别和替换)。
4)将不同样本的输出结果放在同一个文件夹下或同一文件夹的不同子文件夹下,可通过直接下载该文件夹从而下载到所有
的运行结果,省去后续挨个下载运行结果的麻烦(具体见附录)。
(3) 修改参数项:(可选)
1)如果参数值需根据输入的样本发生变动,可参照输入项替换成变量,利用python脚本进行替换(在下文样本文件列表sample.txt中添加相应参数列);
2)如果参数不需要根据输入的样本变动,可直接在模板中修改其值。
(4) 修改任务信息:
1)任务ID:
将id处 < Please input the reference task’s id > 改成null。
2)任务名称:
修改 name 处 < Please input the task’s name in here > :
[1] 可根据 “样本名称+工作流名称” 命名任务,替换成 “{$sample}_Fastqc” ,方便区分不同样本提交的任务;
[2] 也可以自己按命名规则(字母,数字,短横线,下划线)进行命名;
[3] 也可以直接替换成null,系统会自动按该任务所依赖的工作流名加时间戳命名。
(参考模板:Fastqc_param_test.yml(#处为需要修改的地方))
Conditions:
schedule: ''
Inputs:
loaddata_reads:
alias: 输入序列文件
category: loaddata
data:
- enid: null # 输入项enid处修改为null
name: "{$read}" # 输入项name替换成"{$read}",可根据实际情况命名(注意唯一性,方便利用python脚本替换)
property:
block_file:
block_name: null
is_block: false
split_format: default
formats:
- fq
- fastq
- bam
- gz
- sam
maxitems: 1000
minitems: 1
required: true
type: file
Outputs:
store_data_node_fastqc:
alias: store Fastqc_output_tgz
data:
- description: <Please input the description of the output data in here>
name: "/home/admin/Fastqc_Result/{$sample}_fasqtc_out.tgz" # 输出项name替换,可根据实际情况命名(注意唯一性,方便利用python脚本替换)
property:
block_file:
block_name: null
is_block: false
split_format: default
formats:
- tgz
maxitems: 1
minitems: 1
type: file
Parameters:
Fastqc_node1:
alias: fastqc 0.11.3
parameters:
threads:
hint: Specifies the number of files which can be processed simultaneously. Each
thread will be allocated 250MB of memory so you shouldn't run more threads
than your available memory will cope with, and not more than 6 threads on
a 32 bit machine。
maxvalue: 16
minvalue: 1
required: true
type: integer
value: 2 # 参数项根据实际情况修改
variable: true
Property:
CDN:
required: true
reference_task:
- id: null # id处修改为null
water_mark:
required: true
style: null
description: <Please input the task's description in here>
name: "{$sample}_Fastqc" # 修改任务名称
3. 制作样本文件列表
小W:“根据配置文件中需要批量替换的地方,即 “{$..}” 处,制作批量提交的样本文件列表。例如:上述模板中一共有两处 “{$..}”,即 “{$sample}” 和 “{$read}” 需要根据实际的输入信息变动,分别是样本名称和样本数据路径,可参考 sample.txt 文件格式(以 tab 作为分隔符)。”
小F:“嘿嘿,制作这种格式的文件列表对我来说小case一桩,比一个一个地点击加载几百甚至几千个文件省事多了。”
(参考样本文件列表:samples.txt)
# samle_name read_path
NA12878 public:/demo-data/WES-Germline_NA12878_smallsize/NA12878-NGv3-LAB1360-A_1000000_1.fastq.gz
F4-1 biolam_105:/F4-1-tmp_1.fq.gz
F3-10 biolam_105:/F3-10-tmp_1.fq.gz
说明:
对于输出结果比较多的工作流,可以增加输出文件的路径(参照输入项的替换形式)。
4. 批量提交任务
小F:”我在GeneDock的账号是 genedock_f,但我想运行安装的公共工作流 Fastqc(属于 public 账号),这时候该怎么办呢?“
小W:”这时候要理解 res_account_name 的含义,即资源账号。运行任务是在你自己的账号下,所以Python SDK创建 task 对象的时候,res_account_name 是自己的账号,即 genedock_f,但 active_workflow 的时候,由于工作流属于public账号,workflow_owner 为public。当运行自己账号下的工作流时,workflow_owner 可以省略不写,默认值是自己的账号。“
小F:“我准备好配置文件 Fastqc_param_test.yml 和样本文件列表 samples.txt 后,修改 gd_run_task.py 脚本4处需修改的地方,在Shell中执行 python gd_run_task.py 即可批量提交任务吗?“
小W:”是的,简单吧。提交任务的同时,在你当前目录下会生成一个 gd_task.log 日志文件,用于记录批量提交任务时的一些关键信息,便于对任务提交状况进行跟踪。”
小F:“棒棒哒,有了日志记录,如果任务提交失败就可以及时查找到原因了。”
(Python参考脚本2:gd_run_task.py)
#!/usr/bin/env python
# -*- coding: utf-8 -*-
import os
import logging
import gdpy
# 记录任务运行的日志信息,日志信息存储在当前目录下的gd_task.log文件中
LOG_FILENAME = os.path.join(os.getcwd(), 'gd_task.log')
LOG_FORMAT = "[%(asctime)s] - %(filename)s - %(funcName)s - %(lineno)d - [%(levelname)s] : %(message)s"
# 填写秘钥信息,账号信息及ENDPOINT地址(此处需修改-1)
ACCESS_KEY_ID = ''
ACCESS_KEY_SECRET = ''
ENDPOINT = 'cn-beijing-api.genedock.com' # 北京域
RES_ACCOUNT_NAME = '' # 填写自己在GeneDock网站申请的账号
# 工作流名称,版本及所属账号(此处需修改-2)
WORKFLOW_NAME = 'Fastqc'
WORKFLOW_VERSION = 1
WORKFLOW_OWNER = 'public' # 工作流所属的账号,如果是公共工作流,则为public;若属于自己账号,则与RES_ACCOUNT_NAME相同
# 运行任务配置文件和样本文件列表(此处需修改-3)
TEMPLATE_FILE = 'Fastqc_param_test.yml'
SAMPLE_LIST = 'samples.txt'
# 初始化Auth信息
auth = gdpy.GeneDockAuth(ACCESS_KEY_ID, ACCESS_KEY_SECRET)
def init_logger():
"""
初始化记录日志信息
"""
logging.captureWarnings(True)
logging.basicConfig(filename=LOG_FILENAME, format=LOG_FORMAT)
logger = logging.getLogger("GeneDock")
logger.setLevel(logging.INFO)
return logger
_logger = init_logger()
def active_workflow(auth):
"""
创建一个task对象,运行工作流,提交任务,同时处理可能的错误信息
"""
task = gdpy.Tasks(auth, ENDPOINT, RES_ACCOUNT_NAME, connect_timeout=3600)
try:
resp = task.active_workflow(TEMPLATE_FILE + '_tmp', WORKFLOW_NAME, WORKFLOW_VERSION, WORKFLOW_OWNER)
_logger.info('Start task successfully! task_name: {}, task_id: {}'.format(
resp.task_name, resp.task_id))
print('Start task successfully!\ntask_name: {}, task_id: {}'.format(
resp.task_name, resp.task_id))
except gdpy.exceptions.RequestError as e:
_logger.warning('Failed to start task! status: {}, error: {}'.format(
e.status, e.error_message.decode('utf-8')))
print('Failed to start task! status: {}\nerror: {}'.format(
e.status, e.error_message.decode('utf-8')))
print('NetWork Connection Error, please check your network and retry.')
except gdpy.exceptions.ServerError as e:
_logger.warning('Failed to start task! status: {}, error: {}, 错误: {}'.format(
e.status, e.error_message.decode('utf-8'), e.error_message_chs.decode('utf-8')))
print('Failed to start task! status: {}\nerror: {} 错误: {}'.format(
e.status, e.error_message.decode('utf-8'), e.error_message_chs.decode('utf-8')))
if e.status // 100 == 5:
print('Please retry, if failed again, contact with the staff of GeneDock.\n')
elif e.status // 100 == 4:
print('Please check the workflow name/version/owner or template file.\n')
except ValueError as e:
_logger.warning('Failed to start task! error: {}'.format(e))
print('Failed to start task!\nerror: {}'.format(e))
print('Please check the input "workflow name" or "workflow version".\n')
def main():
"""
加载配置文件,根据样本文件列表循环替换配置文件,每次替换完提交一个任务
"""
workflow_template = gdpy.yml_utils.yaml_loader(TEMPLATE_FILE)
for line in open(SAMPLE_LIST):
if line.startswith('#'):
continue
line = line.strip()
ele = line.split('\t') # tab分割
# 根据样本文件列表中相应元素,替换配置文件中需根据样本信息变动的地方,即"{$##}"(此处需修改-4)
workflow_param = str(workflow_template).replace("{$read}", ele[1])
workflow_param = workflow_param.replace("{$sample}", ele[0])
workflow_param = eval(workflow_param)
workflow_param = gdpy.yml_utils.yaml_dumper(workflow_param)
with open(TEMPLATE_FILE + '_tmp', 'w') as f:
f.write(workflow_param.decode('utf-8'))
active_workflow(auth)
# 测试时,可以注释掉下方,取消删除临时文件,便于查看替换后的临时配置文件内容和查找错误原因。
os.remove(TEMPLATE_FILE + '_tmp')
if __name__ == '__main__':
main()
说明:
1)日志文件中记录了任务是否提交成功,提交成功的任务名称和任务ID或提交失败时的错误原因。
(参考日志文件:./gwc.log)
[2017-05-10 16:10:27,755] - task_run.py -
- 67 - [INFO] : Start task successfully. status code: 200; message: {“task_name”: “NA12878_Fastqc”, “task_id”: “5912caf38432cd002f2f1fc9”}
[2017-05-10 16:19:53,444] - task_run.py -
- 82 - [WARNING] : Failed to start task! status: 400, error: Activate workflow failed, since the input is invalid. message: WorkflowNotFound Can’t find workflow. Name: ‘Fastqc22’, version: 1., 错误: 创建任务时给定的输入参数错误.找不到工作流。名称:Fastqc22,版本:1。
2)测试时,可以注释掉gd_run_task.py脚本中 os.remove(TEMPLATE_FILE + ‘_tmp’) 这一行,取消删除临时文件,便于查看替换后的临时配置文件内容以及查找提交任务失败的原因。
3)注意:一次提交较多的人全基因组流程相关的任务时,请提前与GeneDock相关负责人联系。
5. 查看任务列表
小W:“任务提交成功后,就可以登录到GeneDock网站-任务/报告中心,查看你批量提交的任务啦。”
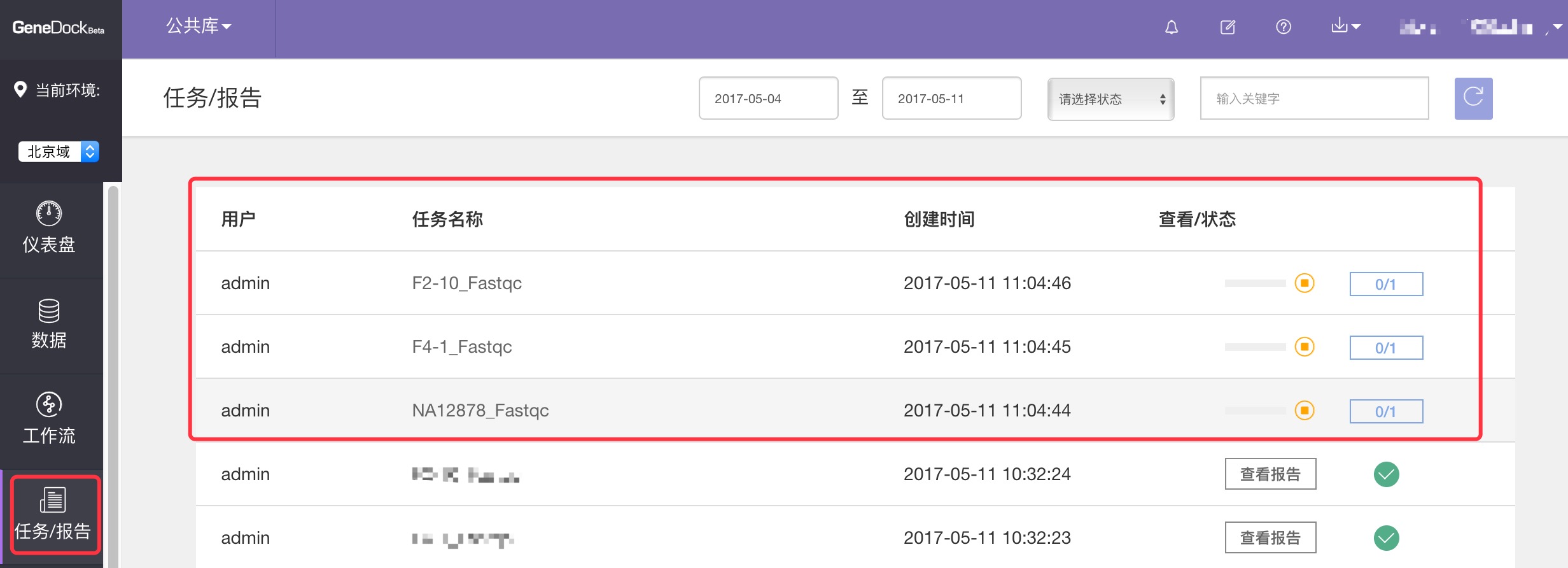
小F:“哇偶,成百上千次分析so easy!主任再也不用担心我们的样本数量问题了。”
小W:“这下又可以受到主任表扬了吧?哈哈。”
附录:
批量下载运行结果
(1) 查看运行结果
任务运行成功后,可以在GeneDock网站-数据中心看到运行结果。
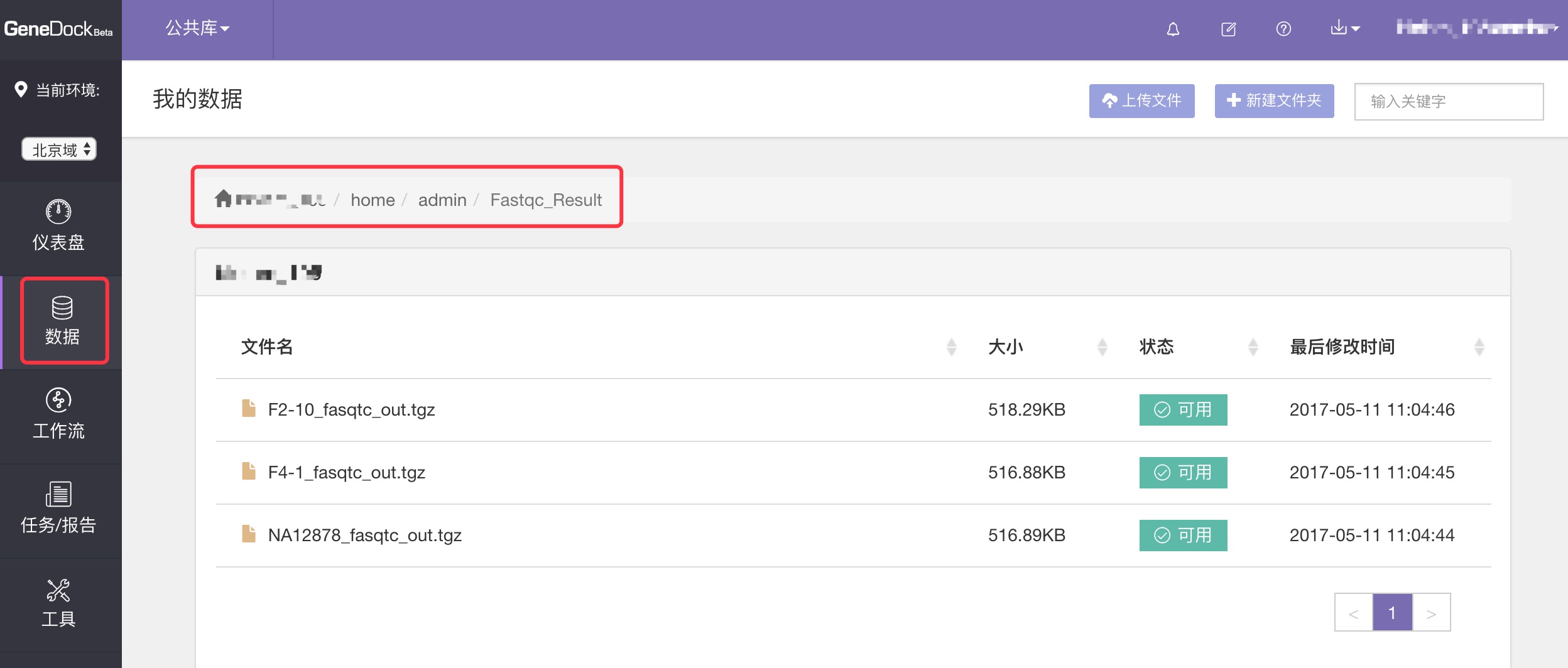
说明:
本例配置文件中,输出项的路径为 “/home/admin/Fastqc_Result/{$sample}_fasqtc_out.tgz”,即将本次批量提交任务的运行结果都输出在了 ”/home/admin/Fastqc_Result/“ 文件夹下。这样设置的一个好处是,方便批量下载运行结果。
(2) 批量下载运行结果
通过命令行/图形化数据客户端批量下载运行结果。本例中,直接下载 “/home/admin/Fastqc_Result/“ 文件夹,就可以把本次任务的所有运行结果下载到本地。参考命令(命令行客户端):
genedock download /account_name/home/admin/Fastqc_Result/(3) 关于数据客户端
数据客户端的下载、安装、配置以及上传和下载数据可以参考GeneDock网站-开发者中心-帮助文档:数据传输工具
相关链接
GeneDock Python SDK:从入门到…放(大)弃(误)?精通!弃(误)?精通!/)
GeneDock Offical Python SDK手册弃(误)?精通!/)