fastq文件格式说明(wiki)
- FASTQ_format 维基百科
- NSC_2011_Illumina_fastqAndQC Illumina fastq 格式官方文档
fastq 文件质量控制
- fastqc
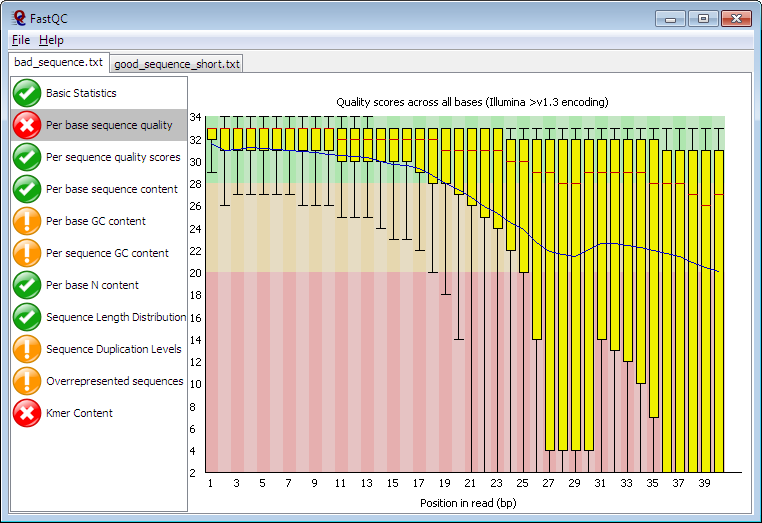
CommandLine Demo:./FastQC/fastqc -o ./ –extract -f fastq -t 4 -q file.fq.gz solexaQA
Dependency:R, gcc, perl

fastx toolkit
Dependency: g++, PerlIO::gzip, GD::Graph::bars, sed, gnuplot- fastx_clipper:
截3’端 adaptor的程序,仅支持单端测序 - fastx_trimmer:
截取位于start-end区间,或者从末端截掉一定长度的序列,输出fastq格式 - fastx_quality_stats:
输出质量值统计结果,可用fastq_quality_boxplot_graph.sh做质量值boxplot图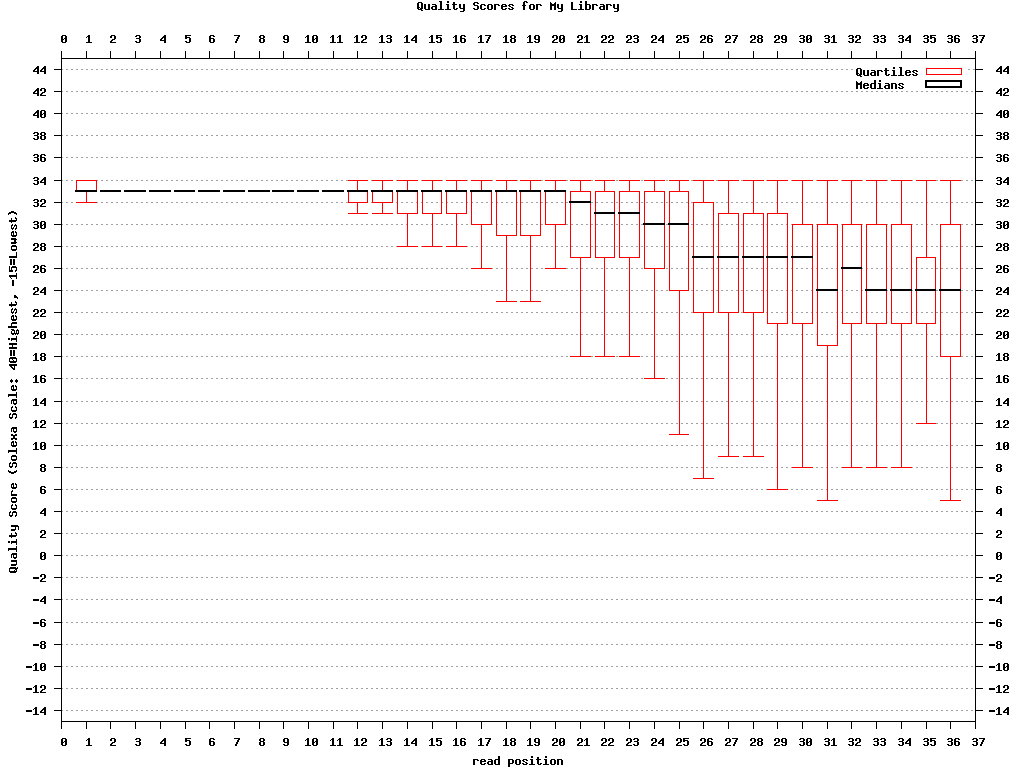
- fastq_to_fasta:
将fastq转换成fasta格式 - fastq_quality_filter:
根据质量值筛选过滤,质量值低于cutoff的将被过滤掉 - fastq_quality_trimmer:
根据质量值截取序列,质量值低的3’ end部分将会被截短,如果截取之后剩余长度不足最小长度阈值,则这条read将会被过滤掉 - fastx_reverse_complement:
取反向互补序列 - fastx_collapser:
输出每条序列及其出现的频数(duplication level) - fastx_uncollapser:
输出文件参数为fastx_collapser的输出文件,将出现多少次的序列输出多少次 - fasta_formatter:
主要要用来控制输出fasta格式文件每行的序列长度 - fasta_nucleotide_changer:
主要用来切换T与U碱基(DNA/RNA)模式 - fastx_renamer:
用read序号数作为read name - fastq_masker:
将read中低于质量值阈值的碱基 mask成其他字符(’.’ or ‘N’) - fastx_nucleotide_distribution_line_graph.sh:
输入文件为fastx_quality_stats的输出结果,用于画所有reads在每个cycle的碱基含量分布线形图 - fastx_nucleotide_distribution_graph.sh:
同上,做stacked barplot图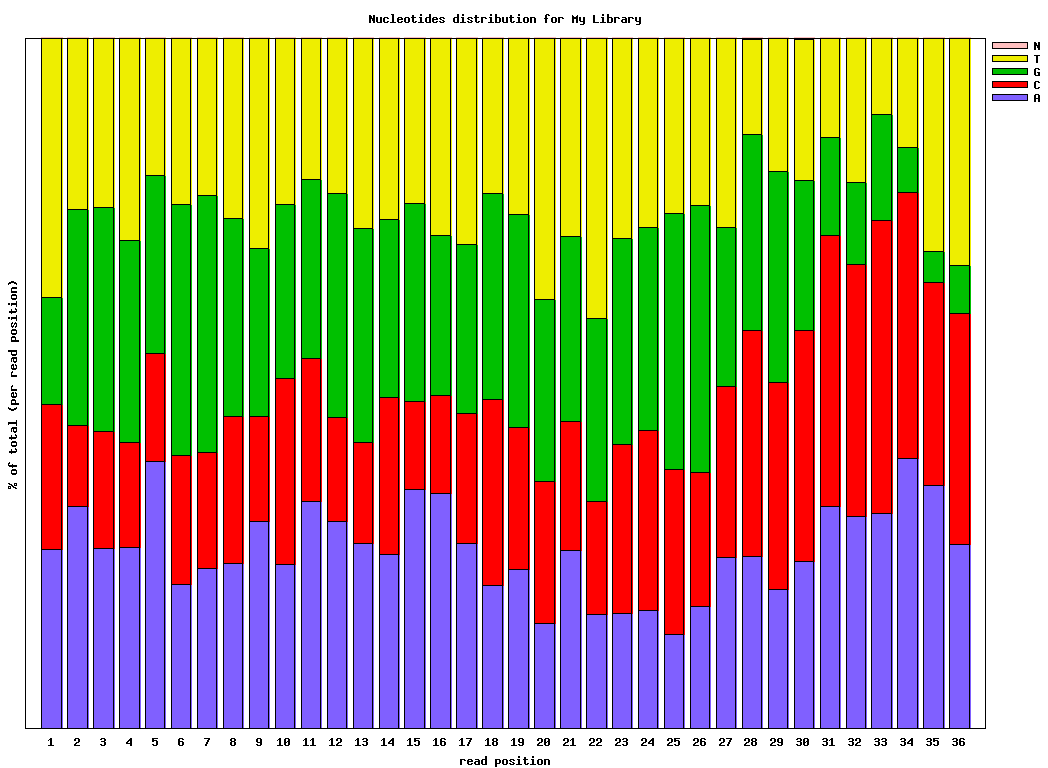
根据fastq文件的5‘end or 3’end 固定长度的序列种类,来分割子文件,可容mismatch容gap - fasta_clipping_histogram.pl:
对fastx_clipper, fastx_trimmer, fastq_quality_trimmer处理的结果做统计,画长度分布图。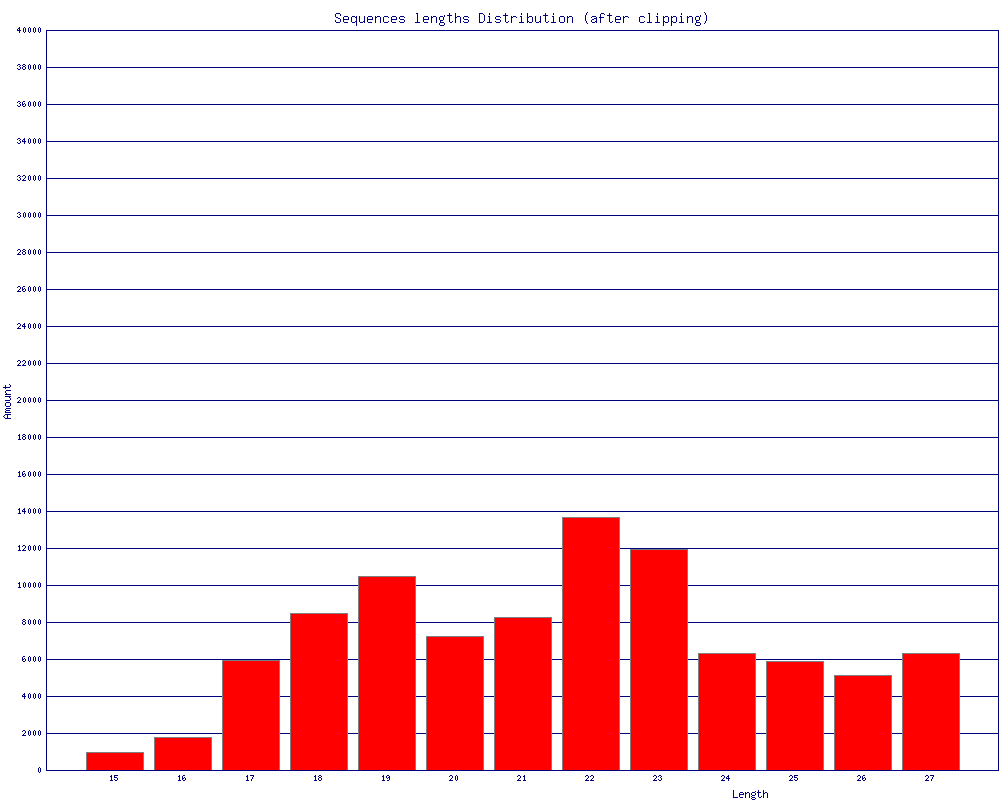
- fastx_clipper:
- fastq-tools
Dependency: gcc,prce- fastq-grep: find reads matching a regular-expression
- fastq-kmers: count k-mer occurances
- fastq-match: local alignment of a sequence to each read
- fastq-sample: randomly sample reads with or without replacement
- fastq-sort: sort fastq files
- fastq-uniq: filter reads with identical sequences
flexbar
http://sourceforge.net/p/flexbar/wiki/Manual/- Description:
flexbar支持多线程 adaptor clipper,支持Pair End,支持 barcode trimmer,支持多种截取模式 Dependency:
CommandLine Demo:
flexbar -n 4 -at 2 -u 1 -m 20 -ao 5 -f i1.8 -a $adaptor --reads $indir/$fastq1 --reads2 $indir/$fastq2 -z GZ -t $outfile_prefix > $outfile_prefix.flexbar.log
- Description:
trimmomatic A flexible read trimming tool for Illumina NGS data
Dependency: javaPair End Demo:
java -jar trimmomatic-0.30.jar PE -phred33 input_forward.fq.gz input_reverse.fq.gz output_forward_paired.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36Single End Demo:
java -jar trimmomatic-0.30.jar SE -phred33 input.fq.gz output.fq.gz ILLUMINACLIP:TruSeq3-SE:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
- seqtk
Dependency: gcc - Fqutils
Dependency: gcc- fqu_are_paired
Checks if two files are associated pairwise: Does the first record appearing in FASTQ file_1 have the same identifier as the first record in FASTQ file_2? …, etc. - fqu_cat
Like Unix cat, but for files in FASTQ format. Assuming well-formed input the output is guaranteed to be in lazy-FASTQ records. Especially useful when your own parsers assume this simpler format. - fqu_cull
Reports a subset of a FASTQ stream, based on a collection of identifiers. - fqu_degen
Replaces A, C, G, T with degenerate codes. Options include conversion to purines and pyrimidines (A, G -> R and C, T -> Y) and to a GC representation (C, G -> S and A, T -> W). - fqu_rselect
Randomly select a subset of records from FASTQ input. - fqu_splitq
Segregate FASTQ input into two output files, based on a collection of identifiers. - fqu_summary
Generate summary statistics for FASTQ input. These include total record (sequence) count, maximum sequence size, a estimation of the quality offset (33 or 64), basecounts per position, a GC content histogram, and per position quartiles of the qualities. - fqu_tidy
Write FASTQ output based on FASTQ input. Like fqu_cat, output is guaranteed to be in four line records. Provides a “–squash” option for reducing the size of each record, currently by ignoring the second header, and a “–tab-delimited” option if you happen to want the output stream in a four column format. These single line records can be particularly convenient for internal processing pipelines. - fqu_wc
Generates word (kmer) counts for FASTQ input.
- fqu_are_paired
- SeqPrep
- homerTools
http://homer.salk.edu/homer/ngs/homerTools.html
http://homer.salk.edu/homer/basicTutorial/fastqFiles.html



fastq压缩
- BEETL-fastq
http://arxiv.org/pdf/1406.4376v1.pdf - fastqz
- scalce
- DSRC
模拟生成fastq格式文件
- XS
-
Program: wgsim (short read simulator) Version: 0.3.0 Contact: Heng Li <lh3@sanger.ac.uk> Usage: wgsim [options] <in.ref.fa> <out.read1.fq> <out.read2.fq> Options: -e FLOAT base error rate [0.020] -d INT outer distance between the two ends [500] -s INT standard deviation [50] -N INT number of read pairs [1000000] -1 INT length of the first read [70] -2 INT length of the second read [70] -r FLOAT rate of mutations [0.0010] -R FLOAT fraction of indels [0.15] -X FLOAT probability an indel is extended [0.30] -S INT seed for random generator [-1] -h haplotype mode
k-mer计数
- khmer
- KMC
- JELLYFISH
genome size estimating based on kmer-counting https://github.com/josephryan/estimate\_genome\_size.pl - BF Counter
Pair End read 连接
-
Usage: fastq-join [options] <read1.fq> <read2.fq> [mate.fq] -o <read.%.fq> Joins two paired-end reads on the overlapping ends. Options: -o FIL See 'Output' below -v C Verifies that the 2 files probe id's match up to char C use '/' for Illumina reads -p N N-percent maximum difference (8) -m N N-minimum overlap (6) -r FIL Verbose stitch length report -R No reverse complement -V Show version Output: You can supply 3 -o arguments, for un1, un2, join files, or one argument as a file name template. The suffix 'un1, un2, or join' is appended to the file, or they replace a %-character if present. If a 'mate' input file is present (barcode read), then the files'un3' and 'join2' are also created. Files named ".gz" are assumed to be compressed, and can be read/written as long as "gzip" is in the path. - COPE
http://bioinformatics.oxfordjournals.org/content/early/2012/10/08/bioinformatics.bts563.full.pdf - PEAR
- FLASH
http://ccb.jhu.edu/software/FLASH/FLASH-reprint.pdffastq文件数据纠错(基于K-mer)
- reptile
Dependency:make, g++, perl
https://code.google.com/p/ngopt/wiki/ReptileErrorCorrection - quake
- LSC for PacBio long read error correction